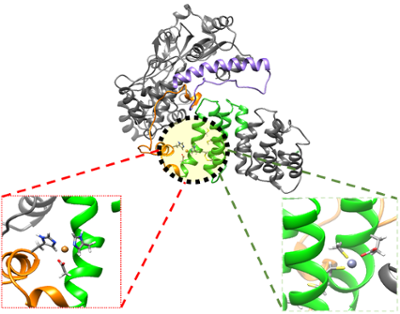
 Huntington’s Disease (HD) is an autosomal dominant genetic disorder characterized by a CAG trinucleotide repeat expansion in the huntingtin protein (htt), giving rise to the mutant form of the protein (mhtt). Critical polyQ tail lengths in the upper 30s have been linked to a novel polyQ conformation that is pathogenic, where patients experience neuronal loss within the striatum and cerebral cortex resulting in mood swings, depression, impaired judgment, involuntary movements, slurred speech and ultimately death. Elevated copper and iron concentrations have also been observed in the striata of HD patients and affected HD transgenic mice, and it has even been suggested that these redox metals may interact directly with and oxidize the polyQ-containing N-terminal fragment. Although the nature of this interaction and subsequent reduction reaction remains unclear, researchers have conclusively shown that accumulation of redox metals is a significant contributing factor to oxidative stress in Huntington’s disease and more broadly to neurodegenerative disease progression. Our group works to provide detailed, metal selective spectroscopic information to define the electronic structure, geometry, ligation, and redox properties of the metal environment in htt and mhtt. Additionally, we utilize biochemical assays, separation methods, and spectroscopic measures for protein secondary structure and stability to aid in elucidating the mechanism by which biorelevant metals contribute to disease progression. All of this is connected to in vivo studies carried out via close collaborations. Our goal is to provide information that can be used to enhance the selectivity of commonly used chelation therapeutics that are administered to reduce the severity of HD symptoms.
Huntington’s Disease (HD) is an autosomal dominant genetic disorder characterized by a CAG trinucleotide repeat expansion in the huntingtin protein (htt), giving rise to the mutant form of the protein (mhtt). Critical polyQ tail lengths in the upper 30s have been linked to a novel polyQ conformation that is pathogenic, where patients experience neuronal loss within the striatum and cerebral cortex resulting in mood swings, depression, impaired judgment, involuntary movements, slurred speech and ultimately death. Elevated copper and iron concentrations have also been observed in the striata of HD patients and affected HD transgenic mice, and it has even been suggested that these redox metals may interact directly with and oxidize the polyQ-containing N-terminal fragment. Although the nature of this interaction and subsequent reduction reaction remains unclear, researchers have conclusively shown that accumulation of redox metals is a significant contributing factor to oxidative stress in Huntington’s disease and more broadly to neurodegenerative disease progression. Our group works to provide detailed, metal selective spectroscopic information to define the electronic structure, geometry, ligation, and redox properties of the metal environment in htt and mhtt. Additionally, we utilize biochemical assays, separation methods, and spectroscopic measures for protein secondary structure and stability to aid in elucidating the mechanism by which biorelevant metals contribute to disease progression. All of this is connected to in vivo studies carried out via close collaborations. Our goal is to provide information that can be used to enhance the selectivity of commonly used chelation therapeutics that are administered to reduce the severity of HD symptoms.

