Abstract:
Cyclic carbonate is an important intermediate for preparing polycarbonates, which are crucial materials in diverse research fields. This study introduced a three-step approach to synthesizing a disulfide-containing cyclic carbonate monomer, with confirmation through proton nuclear magnetic resonances (1H NMR) and high-resolution mass spectrum analysis. The resulting monomer holds the potential for subsequent synthesis of responsive functional aliphatic polycarbonates, providing promise for advanced biomedical applications.
Introduction:
Aliphatic polycarbonates represent a significant compound category that serves crucial roles in various biological contexts.1 These synthetic polymers offer the advantage of simplified handling and excellent reproducibility. They find applications in diverse areas like polymer-drug conjugates, drug encapsulation, and polymeric contrast agents for imaging.1,2 The ring-opening polymerization of cyclic carbonate monomer is an important strategy for the synthesis of these polycarbonates. A long-standing goal of our project is to achieve stimuli-responsive drug delivery in cancer cells based on the glutathione-responsive disulfide bonds. Toward this end, we tried to use a simple and efficient method to prepare a disulfide-containing cyclic carbonate monomer for further biomedical applications (Scheme1).
Experimental Details:
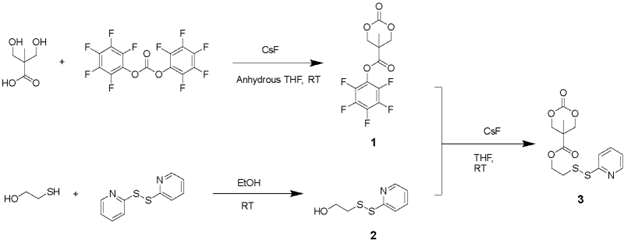
Scheme 1. Synthesis route of the disulfide-containing cyclic carbonate monomer
Synthesis of Compound 1
2,2-Bis(hydroxymethyl)propionic acid (bis-MPA) (1.50 g, 11 mmol), bis-(pentafluorophenyl)carbonate (PFC) (10.90 g, 55 mmol, 2.5 eq.), and CsF (0.35 g, 2.3 mmol, 0.2 eq.) were dissolved in 40 mL of anhydrous tetrahydrofuran (THF) in a 100 mL round bottom flask. The homogeneous solution was stirred for 24 hours at room temperature. After removing the solvent under vacuum, the residue was re-dissolved in CH2Cl2, and the filtrate was extracted with saturated NaHCO3 solution and NaCl solution, respectively. The organic layer was dried with Na2SO4, and then evaporated in vacuum. The crude product was recrystallized from ethyl acetate/hexane mixture (1/1, v/v) to give the compound 1 as a white crystalline powder (1.87 g, yield = 51.3%). 1H NMR (400 MHz, CDCl3-d) δ: 4.86 (d, J = 8 Hz, 2H), 4.37 (d, J = 8 Hz, 2H), 1.57 (s, 3H) ppm. HRMS (ESI) m/z: [M+Na]+ calculated for C12H7O5F5Na 349.0106, found 349.0098; [2M+Na]+ calculated for C24H14O10F10Na 675.0325, found 675.0309.
Synthesis of Compound 2
2-Mercaptoethanol (1.0 g, 12.8 mmol) and 2,2’-dipridyldisulfide (4.23 g, 19.2 mmol, 1.5 eq.) were dissolved in 20 mL of ethanol. The solution was stirred for 24 hours at room temperature. After removing the solvent under reduced pressure, the residue was purified with silica gel chromatography (hexane/ethyl acetate = 3/1 to 1/1 as the eluent) to give compound 2 as the oil (570 mg, yield = 47%). 1H NMR (400 MHz, CDCl3-d) δ: 8.50 (d, J = 4 Hz, 1H), 7.58 (t, J = 8 Hz, 1H), 7.41 (d, J = 8 Hz, 1H), 7.14 (t, J = 8 Hz, 1H), 5.07 (br, 1H), 3.80 (t, J = 4 Hz, 2H), 2.95 (t, J = 6 Hz, 2H) ppm. HRMS (ESI) m/z: [M+Na]+ calculated for C7H9NOS2Na 210.0023, found 210.0019.
Synthesis of Compound 3
Compound 1 (0.5 g, 1.53 mmol), CsF (109 mg, 0.72 mmol, 0.47 eq.), and Compound 2 (327 g, 1.75 mmol, 1.14 eq.) were dissolved in 20 mL of anhydrous THF. The mixture was stirred for 24 hours and then filtered to remove pentafluorophenol byproduct. After evaporating the solvent in a vacuum, the reaction mixture was re-dissolved in CH2Cl2. Then the solution was allowed to stand for 30 min and filtered again to remove additional precipitated pentafluorophenol. The organic solution was washed with saturated NaHCO3 and NaCl (200 mL) aqueous solution and then dried over Na2SO4. The concentrated crude product was purified by silica gel column chromatography (ethyl acetate/hexanes = 1/1 to 2/1 as the eluent) to obtain compound 3 as an oil product (270 mg, yield = 53%). 1H NMR (400 MHz, CDCl3-d) δ: 8.47 (d, J = 8 Hz, 1H), 7.65 (m, 2H), 7.12 (d, J = 4 Hz, 1H), 4.68 (d, J = 12 Hz, 2H), 4.47 (t, J = 8 Hz, 2H), 4.20 (d, J = 8 Hz, 2H), 3.07 (t, J = 6 Hz, 2H), 1.33 (s, 3H) ppm. HRMS (ESI) m/z: [M+H]+ calculated for C13H16NO5S2 330.0470, found 330.0492.
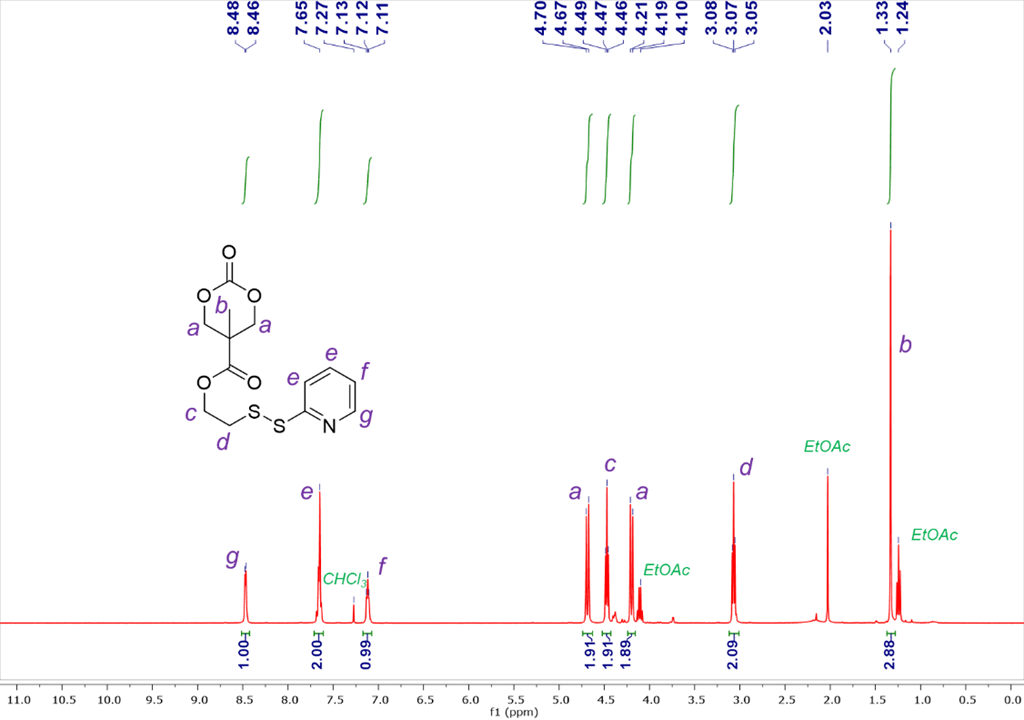
Figure 1. 1H NMR for Compound 3
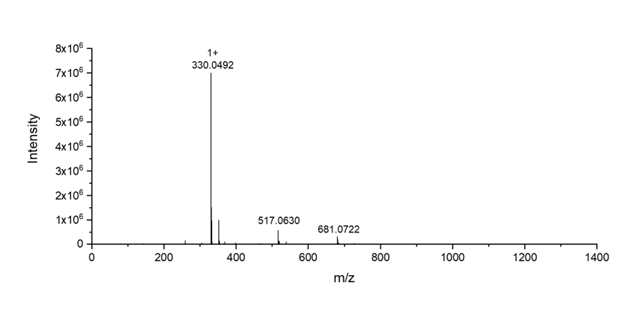
Figure 2. High Resolution Mass Spectrum for compound 3. [M+H]+ calculated for C13H16NO5S2 330.0470, found 330.0492.
Results and discussion:
We successfully synthesized a disulfide-containing cyclic carbonate monomer in three steps from commercially available compounds with a high purity and good yield (Scheme 1). Compound 1 was synthesized from bis-MPA and PFC under the catalyst of CsF. They were dissolved in THF and reacted at room temperature for 24 h to yield compound 1 as a white crystallized powder in 51% yield. Compound 2 was synthesized from 2-mercaptoethanol and 2,2’-dipridyldisulfide in ethanol. The resulting compound 2 was purified by silica gel column chromatography to produce an oil with a 47% yield. Finally, compound 3 was produced by the reaction between compound 1 and compound 2 in THF. After reacting at room temperature for 24 h, the byproduct was removed by extraction, precipitation, and silica gel column chromatography to obtain compound 3 as an oil product in 53% yield. All the compounds have been characterized by 1H NMR and high-resolution mass spectrometry (Figures 1 and 2).
Conclusion:
We used three steps to synthesize a functionalized cyclic carbonate monomer under mild reaction conditions in a high yield. The cyclic carbonate monomer will be further utilized for ring-opening polymerization to produce stimuli-responsive aliphatic polycarbonates for drug delivery.
Acknowledgements:
This work is supported by the National Science Foundation CAREER award under grant No. 2238812.
Eva Bukhryakova expresses her gratitude to the Zhang Research Group for their exceptional support.
References:
- Wei Yu, Edward Maynard, Viviane Chiaradia, Maria C. Arno, and Andrew P. Dove, Aliphatic Polycarbonates from Cyclic Carbonate Monomers and Their Application as Biomaterials, Chemical Reviews, 2021, 121 (18), 10865-10907.
- Daniel P. Sanders, Kazuki Fukushima, Daniel J. Coady, Alshakim Nelson, Masaki Fujiwara, Manabu Yasumoto, and James L. Hedrick, A Simple and Efficient Synthesis of Functionalized Cyclic Carbonate Monomers Using a Versatile Pentafluorophenyl Ester Intermediate, Journal of the American Chemical Society, 2010 132 (42), 14724-14726

