Abstract
Peptides provide promising building blocks to fabricate adaptable self-healing molecular constructs. Recent advancements in the area provide details about the peptides that are capable of catalyzing several reactions with remarkable activities. In this project, aggregation of peptides with different sequences of amino acids has been investigated to understand their structural properties.
Introduction
Peptides exhibit remarkable self-assembly capabilities, forming diverse structures with a wide range of physical characteristics and properties. This self-assembly occurs through non-covalent interactions like Van der Waals forces, hydrogen bonding, electrostatic interactions, and stacking. One application lies in the development of catalytic hydrogels through peptide modification. Within these self-assembling peptide hydrogels, specific amino acid sequences can be designed to mimic the active sites of natural enzymes, granting them enzyme-like activity for catalyzing various chemical reactions. By incorporating catalytically active peptide sequences, our research aims to achieve enhanced catalytic efficiency and selectivity, akin to natural enzymes. Moreover, the tunability of peptide hydrogels allows for the design of custom catalysts for specific reactions, making them versatile tools for organic synthesis and other chemical processes. Although synthetic catalysts have yet to match the efficiency of enzymes, there is a growing demand for catalysts that mimic enzymatic properties in non-essential reactions within the pharmaceutical industry. These peptide chains can act as catalysts by providing specific binding sites, stabilizing transition states, showing enantioselectivity, and offering easy synthesis, thereby making them versatile and valuable tools in catalysis.
In this project, our focus was on modifying various heterogeneous peptides and investigating their reactions with and without zinc. We used ALF to substitute its natural counterpart, histidine, due to its distinct bridging and aggregation properties between chains. This substitution also introduced flexibility in the receptor site, enabling the use of heavier metals. Valine was introduced to maintain a hydrophobic core, crucial for the self-healing properties of the resulting structure, while arginine and asparagine, charged amino acids, reinforced the self-healing properties.
Through computational tools like GROMACS and YASARA we were able to modify and build these structures. The molecules created underwent molecular dynamic simulations, which resolved atomic movement equations to calculate the equilibrium and transport properties of the systems. Such simulations are particularly valuable in medicinal research, aiding in the prediction of protein-ligand docking and understanding physical interactions within molecules under standard temperature and pressure (293.15K & 14.696 psi), leading to equilibrated structures.
Computational Details
All molecular dynamics (MD) simulations conducted utilizing the GROMACS 2022 software package. The AMBER 99sb force field was employed to describe the interactions and behavior of the simulated systems during the MD simulations. For two different peptide lengths, 7-peptide and 9-peptide, MD simulations were performed for a duration of 100 nanoseconds each, allowing for an extensive calculation of the peptide dynamics. In the case of simulations involving zinc, a pre-equilibrated structure was utilized as the starting point, and MD simulations were carried out for a duration of 50 nanoseconds. This approach aimed to investigate the behavior of the system in the presence of zinc and understand its effects over a shorter timescale. Parameters necessary for the simulation of the α-lipoic acid (ALF) molecule were calculated using the ANTECHAMBER module from Gaussian output. This step ensured accurate parameterization for ALF to be compatible with the simulation environment. For the optimization of the ALF molecule's structure, quantum mechanical calculations were performed using the Gaussian software package. The B3LYP functional, known for its balanced treatment of molecular properties, was employed for these calculations. A basis set of 6-31g(d) was utilized to capture the electronic structure of the ALF molecule effectively during the optimization process.
These computational procedures were meticulously carried out to obtain reliable and meaningful insights into the dynamics and behavior of the studied systems, thereby contributing to the overall understanding of the research objectives.
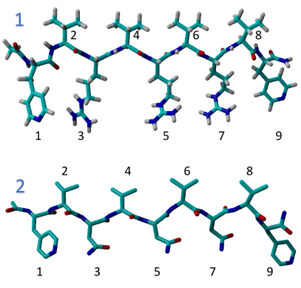

Fig 1. Computational model of the 9 peptide chains:
1. ALF-VRVRVRV-ALF
2. ALF-VNVNVNV-ALF
Results and Discussion
This research aimed to explore the self-healing properties exhibited by peptides featuring alternating hydrophilic and hydrophobic amino acids. These properties were harnessed by designing peptide sequences to elicit the desired outcomes.
To establish a hydrophobic core essential for effective self-healing, the amino acid valine was incorporated into the peptide chain. Valine's hydrophobic nature facilitated the formation of a stable core within the resulting structure, contributing to the self-healing attributes. In parallel, the self-healing properties were further reinforced through the incorporation of arginine, an amino acid carrying a charge. By introducing arginine into the peptide chain, the self-healing properties were enhanced, creating an interplay between the hydrophilic and hydrophobic segments of the sequence.
A second variant of the peptide was also studied, involving the substitution of arginine with asparagine. Asparagine, being another hydrophilic amino acid, offered an alternative perspective on the interplay between hydrophilicity and hydrophobicity within the peptide structure. To investigate the effects of metal binding on aggregation, specific metal binding residues like histidine were studied. In this study, histidine was substituted by α-lipoic acid (ALF). ALF’s distinctive bridging and aggregation capabilities between peptide chains made it an appropriate replacement, enabling the exploration of metal-mediated interactions.
The substitution of ALF for histidine allowed increased flexibility at the receptor site, making it possibly to incorporate heavier metals. By incorporating ALF residues within the peptide chain, a receptor site was engineered to facilitate metal binding interactions.
A) 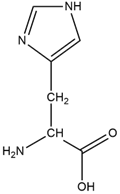 B)
B) 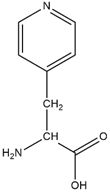
Fig 2. Structure A is the chemical structure for histidine. Structure B is the chemical structure for ALF.
The way in which we arranged the arginine and valine amino acids in the first peptide chain, created strong connections between the individual chains through hydrogen bonds. The valine amino acids were important in blocking water molecules from getting in and interacting with the hydrophilic amino acids, which resulted in a twisted structure that can bounce back. We also looked at an alternative configuration where we used asparagine instead of arginine. In the second peptide chain, the presence of a second carbonyl group in asparagine introduced a set of hydrogen bonding interactions among asparagine residues within the peptide chain. As a result, the interactions between the chains were enhanced, creating an environment conducive to self-association and self-healing.
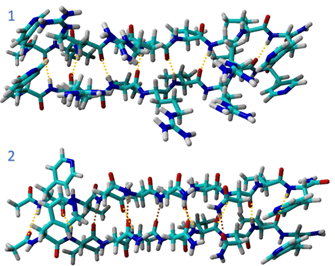
Fig 3. Computational model of the interactions between chains.
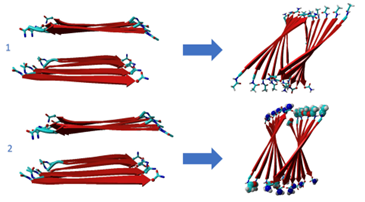
Fig 4. Input and output structure of ALF-VRVRVRV-ALF peptide. Input and output structure ALF-VNVNVNV-ALF peptide.
Conclusion
This research delved into the manipulation of peptides to create adaptable self-healing structures with catalytic potential. Through careful amino acid selection, including the incorporation of ALF, we engineered peptides with robust structures and metal-binding abilities. Computational simulations using GROMACS and YASARA provided crucial insights.
Two peptide variants—one with arginine and one with asparagine—showcased the interplay of hydrophilic and hydrophobic elements, yielding distinct self-assembling structures. Valine reinforced stability by creating a hydrophobic core.
This study advances the field of self-healing molecular design, offering potential for catalysis in organic synthesis and other applications. The extent of twisting and interaction between neighboring chains are different for structures 1 and 2. Further structural studies are required to determine the extent of the differences.
The influence of sequencing in structure and activity is to be explored further.
Equilibrating metal bound peptide structures is the next step of the project.
Active sites with different metals and their activities are important facets of this project, how metals affect the overall aggregation and activity is to be explored further.
Acknowledgements
I extend my gratitude to the Department of Chemistry at the University of Miami for their invaluable collaboration with ACS’s Project Seed. I am sincerely thankful to Project Seed for granting me this opportunity to engage in meaningful research. Lastly, I would like to express my appreciation to Dr. Rajeev Prabhakar and his dedicated graduate students for their mentorship, which greatly enhanced my knowledge of computational chemistry.
References
- D’Souza, A., Marshall, L. R., Yoon, J. H., Kulesha, A., Edirisinghe, D. I. U., Chandrasekaran, S., Rathee, P., Prabhakar, R., & Makhlynets, O. V. (2022). Peptide hydrogel with self-healing and redox-responsive properties. Nano Convergence, 9(1). doi.org/10.1186/s40580-022-00309-7
- D’Souza, A., Yoon, J. H., Beaman, H., Gosavi, P. M., Lengyel-Zhand, Z., Sternisha, A., Centola, G., Marshall, L. R., Wehrman, M. D., Schultz, K. M., Monroe, M. B. B., & Makhlynets, O. V. (2020). Nine-Residue peptide Self-Assembles in the presence of silver to produce a Self-Healing, cytocompatible, antimicrobial hydrogel. ACS Applied Materials & Interfaces, 12(14), 17091–17099. doi.org/10.1021/acsami.0c01154
- Marshall, L. R., Jayachandran, M., Lengyel-Zhand, Z., Rufo, C. M., Kriews, A., Kim, M. C., & Korendovych, I. V. (2020). Synergistic interactions are prevalent in catalytic amyloids. ChemBioChem, 21(18), 2611–2614. doi.org/10.1002/cbic.202000205
- Song, R., Wu, X., Xue, B., Yang, Y., Huang, W., Zeng, G., Wang, J., Li, W., Cao, Y., Wang, W., Lu, J., & Dong, H. (2018). Principles governing catalytic activity of Self-Assembled short peptides. Journal of the American Chemical Society, 141(1), 223–231. doi.org/10.1021/jacs.8b08893
- Zozulia, O., Dolan, M. A., & Korendovych, I. V. (2018). Catalytic peptide assemblies. Chemical Society Reviews, 47(10), 3621–3639.

