Abstract
Cancer-specific ligand-receptor interactions have potential to reveal new information about cancer therapeutics and candidates for treatment. Currently, discovery and classifications of unknown interactions is time-consuming and tedious. This study proposes a novel approach utilizing a random forest algorithm to automate the prediction and classification of ligand-receptor interactions. The chosen algorithm performs strongly, with AUC-PR = 0.79 and MCC = 0.45, yielding innovation in drug discovery for cancer treatment.
Introduction
In cancer-specific compound profiling, ligand-target interactions, the most notable of which being ligandreceptor interactions, have high potential to reveal cancer therapeutics. Indeed, these ligand-receptor interactions have become of high interest to computational and cheminformatics methods to classify and discover treatments for notable cancers, such as lung cancer adenocarcinomas.
Existing methods to investigate these interactions such as virtual screening utilize repetitive clinical trials of compound tests against libraries of targets to provide records of interactions, which are analyzed and organized to reveal potentially effective drugs for disease-related treatments. However, these processes often produce many unknown values for compounds/targets which are more complex, while also requiring significant amounts of time and effort. Prediction and completion of these matrices can be automated by applying machine learning algorithms which process non-linear inputs and classify the treatment potential of drugs. While prior machine learning approaches have been explored in predicting compound profiling matrices, these approaches are primitive with respect to cancer-specific treatments and are unable to properly process complex ligand-receptor interactions.
This study proposes a novel approach to predicting and completing profile matrices for ligand-receptor interactions through the application of a random forest algorithm. This algorithm was trained and validated on data curated from the CancerSCEM database, which captures a variety of ligand-receptor interactions for various cancer samples. The algorithm performed effectively in predicting interactions, with an AUCPR score of 0.79 and an MCC score of 0.45. This study has significant potential to revolutionize methods of computationally drug discovery and yield new compounds that are effective in combating cancer through treatment.
Experimental Details
Data for the model was produced based on curated data from CancerSCEM, an online cancer scRNAseq dataset that concatenates various forms of cancer data from human samples, specifically cell interaction networks. This data contained about 347 various compounds and 1,048 targets for cancer therapy, the known and unknown combinations of which provided the learning base for the algorithm. The target-ligand interactions that were provided on the CancerSCEM database were primarily used as the training data, while the unknown interactions comprised the training set. Features for model development included the scale complexity of the compound involved, chemical makeup, reactivity analysis, and existing trials with similar targets. The sole output was the classification of the interaction between the ligand-receptor pair based on the scale used by CancerSCEM.
Multiple algorithms were trained and tested in order to find the best performing algorithm that would serve as a flagship model for the study. These models were at first trained on the dataset, and was validated using 10-fold cross validation in order to improve the strength and precision of the model. Validation thus ensured that the entirety of the interaction network and the implicit biases between each interaction were wholly considered in output generation.
Performance on the testing set was determined by two factors: Area under the Precision-Recall Curve, which gives a general score for precision and strength, and the Matthew Correlation Coefficient, a higher value of which indicates power in predicting underrepresented outcomes.
Results & Discussion
The algorithm that had the strongest performance with the given metrics proposed by the study was the random forest algorithm, which utilized multiple decision trees to concatenate the data into a single output. This algorithm had a strong AUC-PR score of 0.79 (0 to 1) and a similarly strong and positive MCC score of 0.45 (-1 to +1). Other algorithms with notable performance include the neural network deep learning module, which yielded a AUC-PR score of 0.58 and a stronger MCC score 0.47. The relative strength of the random forest algorithm in accurately predicting the unknown ligand-receptor interactions is of significant importance as it outpaces current advancements and industry standards within the cheminformatics field.
Conclusion
This study proposes a novel method by which to accurately predict and classify the ligand-target interactions for cancer-specific treatments and their target proteins and molecules. Machine learning is effective in this prediction, and the random forest model selected effectively addresses the current problems faced in the pace of innovation in drug treatments. Indeed, this algorithm has strong potential to revolutionize drug discovery by reducing both the time and effort required for novel discoveries in the interactions of compounds and targets, allowing for the development of cancer therapeutics at a faster rate than ever before.
Figures and Captions
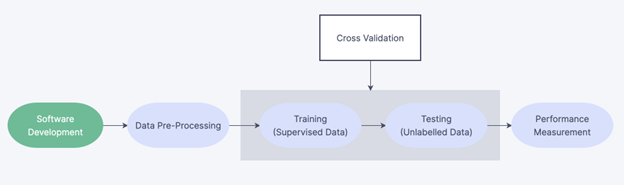
Figure 1: Model development process. All algorithms developed underwent software engineering, data
processing, validation (consisting of training and testing), and a final performance measurement. This
methodology was followed for all models tested in the study.
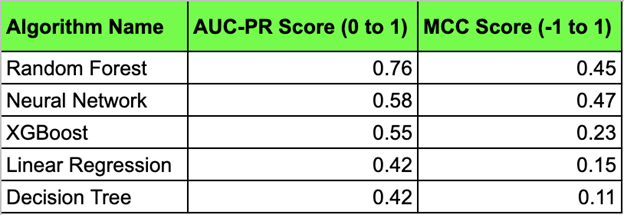
Figure 2: Table capturing the scores of Area Under Precision-Recall Curve and Matthew Correlation
Coefficient for each algorithm tested on the CancerScem dataset. Algorithms were tested and trained with
10-fold cross validation and are ranked from top to bottom based on performance across both scores.
Acknowledgements
I’d like to acknowledge Dr. Malabika Devgan of UMass Memorial for supervision throughout the research
process, as well as funding for the tools and equipment used to conduct this research.
References
- Zhang Y, Song J, Zhang X, Xiao Y. LIGAND-RECEPTOR INTERACTIONS AND DRUG DESIGN.
Biochem Insights. 2015 Dec 20;8(Suppl 1):21-3. doi: 10.4137/BCI.S37978. PMID: 26715850;
PMCID: PMC4687977.
- Chen Z, Yang X, Bi G, Liang J, Hu Z, Zhao M, Li M, Lu T, Zheng Y, Sui Q, Yang Y, Zhan C, Jiang
W, Wang Q, Tan L. Ligand-receptor interaction atlas within and between tumor cells and T cells
in lung adenocarcinoma. Int J Biol Sci. 2020 May 18;16(12):2205-2219. doi: 10.7150/ijbs.42080.
PMID: 32549766; PMCID: PMC7294944.
- Mizera M, Latek D. Ligand-Receptor Interactions and Machine Learning in GCGR and GLP-1R
Drug Discovery. Int J Mol Sci. 2021 Apr 14;22(8):4060. doi: 10.3390/ijms22084060. PMID:
33920024; PMCID: PMC8071054.
- Vogt M, Jasial S, Bajorath J. Computationally derived compound profiling matrices. Future Sci
OA. 2018 Jul 24;4(8):FSO327. doi: 10.4155/fsoa-2018-0050. PMID: 30271615; PMCID:
PMC6153460.
- Rodríguez-Pérez R, Miyao T, Jasial S, Vogt M, Bajorath J. Prediction of Compound Profiling
Matrices Using Machine Learning. ACS Omega. 2018 Apr 30;3(4):4713-4723. doi:
10.1021/acsomega.8b00462. PMID: 30023899; PMCID: PMC6045364.
- Hayes CJ, Dowling CM, Dwane S, McCumiskey ME, Tormey SM, Anne Merrigan B, Coffey JC,
Kiely PA, Dalton TM. Extracellular matrix gene expression profiling using microfluidics for
colorectal carcinoma stratification. Biomicrofluidics. 2016 Oct 31;10(5):054124. doi:
10.1063/1.4966245. PMID: 27822332; PMCID: PMC5097046.
- Zhou JX, Taramelli R, Pedrini E, Knijnenburg T, Huang S. Extracting Intercellular Signaling
Network of Cancer Tissues using Ligand-Receptor Expression Patterns from Whole-tumor and
Single-cell Transcriptomes. Sci Rep. 2017 Aug 18;7(1):8815. doi: 10.1038/s41598-017-09307-w.
Erratum in: Sci Rep. 2018 Dec 12;8(1):17903. PMID: 28821810; PMCID: PMC5562796.
- Jingyao Zeng, Yadong Zhang, Yunfei Shang, Jialin Mai, Shuo Shi, Mingming Lu, Congfan Bu,
Zhewen Zhang, Zaichao Zhang, Yang Li, Zhenglin Du, Jingfa Xiao, CancerSCEM: a database of
single-cell expression map across various human cancers, Nucleic Acids Research, Volume 50,
Issue D1, 7 January 2022, Pages D1147–D1155, https://doi.org/10.1093/nar/gkab905

