Abstract
Peptides are fundamental for the formation of proteins. There are 20 amino acids, and each amino acid sequence makes a peptide. Peptides self-aggregate through different intermolecular forces to make a protein. In this research report, I am aiming to analyze the structures of the RYRY polar proteins sequences compared to the RDRD polar protein, as well as the residue sequences.
Introduction
The elements in my protein are Carbon, Hydrogen, Oxygen, and Nitrogen. The protein that I used consisted of the following amino acids PHE, HIS, TYR, and ARG. Phenylalanine is crucial to produce neurotransmitters such as dopamine and produce the pigment melanin. Histidine is found in many rich proteins such as eggs, fish, and beans. Tyrosine is what helps give you an abundance of adrenaline in a stressful situation. Arginine is part of what produces T-helper cells and regulates your blood flow. The charge in my protein is balanced with an NH3+ added to the first residue and a COO- added to the last residue sequence. With the addition of Na and Cl ions, the protein was able to form ionic bonds. I created 6x2 and 12x2 protein residues.
|
1
|
PHE
|
|
|
2
|
HIS
|
|
3
|
PHE
|
|
4
|
HIS
|
|
5
|
PHE
|
|
6
|
ARG
|
|
7
|
PHE
|
|
1
|
PHE
|
|
|
2
|
HIS
|
|
3
|
PHE
|
|
4
|
HIS
|
|
5
|
PHE
|
|
6
|
TYR
|
|
7
|
PHE
|
|
1
|
PHE
|
 |
|
2
|
HIS
|
|
3
|
PHE
|
|
4
|
HIS
|
|
5
|
PHE
|
|
6
|
ARG
|
|
7
|
PHE
|
|
1
|
PHE
|
 |
|
2
|
HIS
|
|
3
|
PHE
|
|
4
|
HIS
|
|
5
|
PHE
|
|
6
|
ASP
|
|
7
|
PHE
|
Computational Details
Through the application of the computer software GROMACS with the Force Field CHARMM, Molecular Dynamic MD (Simulations) were able to run for 200 ns. Then, I added ions to neutralize solvent and to maintain similar conditions as blood. I was able to minimize the energy of the input system while applying temperature, volume, and pressure. Lastly, I ran a molecular dynamic simulation while doing G-cluster to calculate the average structure.
Results and Discussion
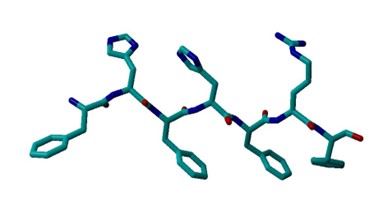
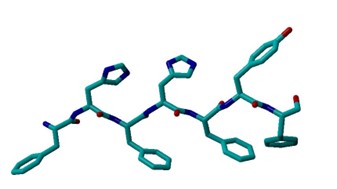
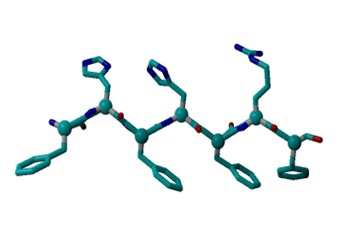
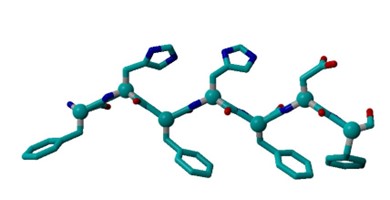
Upon looking at a PHE structure, I see that it forms a hexagonal planar shape with benzyl ring. Tyrosine forms a 4-hyrdoxphenyl shape with 6 carbons and an OH group. Histidine has a pentagonal shape consisting of 3 carbons and 2 nitrogens. Arginine consists of two NH’s bonded to carbon.
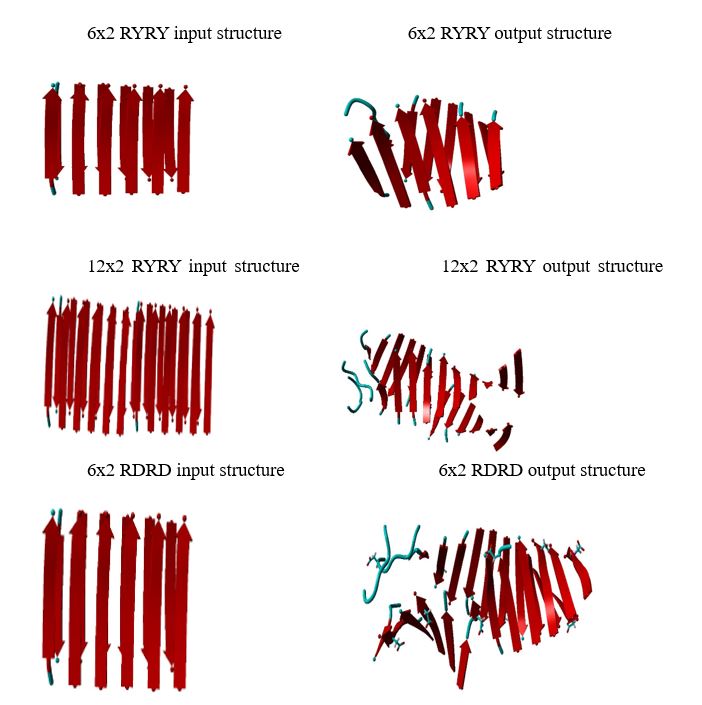
The input files were linear, and the residues were closer together compared to the compared to the more twisted output files. In addition, the 12x2 output structure was more twisted compared to the 6x2 output structure. Furthermore, in the output files, there is hydrogen attached to a Histidine. In the protein structure with Tyrosine, there is an additional OH group and more C-C bonds compared to the protein structure with Arginine.
All of the input files had approximately a marked angle of 75 and a marked dihedral of 5.3. The 6x2 output file with a marked angle of approximately 80 and a marked dihedral of approximately 49.
Residue Sequences
FHFHFRF
FHFHFYF
FHFHDY
Conclusion
In conclusion, peptides play a crucial role in the synthesis of proteins. Through chemical interactions, peptides can self-aggregate and form a protein. The protein can potentially play a role in cell communication, enzymes, hormones, and immune responses. Due to time constraints, I was only able to do preliminary research on the peptides. Future research would include an analysis of the peptides’ properties and interactions with other proteins. Based on the results and discussions, the peptides became more twisted after the molecular dynamic simulations.
Acknowledgements
I want to thank the Chemistry Department of the University of Miami along with Professor Prabhakar and his team for allowing me to do research in their lab. Furthermore, I appreciate the American Chemical Society giving me the opportunity to be a part of this summer internship.
References
- Abraham, M.J.; Murtola, T.; Schultz, R.; Pall, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1-19-25.
- Huang, J.; MacKerell Jr., A.D. CHARMM36 all atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem 2013, 34, 2135-2145

